Chemical space synthesis
Author: Alfredo Quevedo (aquevedo@unc.edu.ar) - July, 2025.
Objective: Create a custom chemical space using combinatorial synthesis
In this example we are going to implement the steps required to reproduce the construction of a virtual chemical space as reported by Cerutti et al in the search of targeted covalent inhibitors (TCI) with antichagasic activity.
The execution of a virtual drug screening campaing encompasess diverse medicinal chemisty objectives, with each project constituting a unique scientific scenario linked to a synthetic strategy.
In this example, the aim of the work was to design potential triazole-based TCI based targeting Cruzipain (CZP), a well-known therapeutic target for treatment of Chagas disease.
The catalytic domain of CZP consists of a single polypeptide chain of 215 aminoacid (AA) residues, forming α-helices and antiparallel β-sheets that fold in two distinct sub-domains, delineating the active site that is located within the interface (Figure 1). This site includes a catalytic triad (Cys25, His162 and Asn182) accompanied by a oxyanion hole (OAH: Gln19 and Cys25), both of which constitutes key structures involved in the mechanism related to the enzyme hydrolytic activity. Extensive structural, mechanistic, and topological studies have been conducted on CZP substrates and inhibitors interacting within these subsites, with structure-activity relationships (SAR) been explored in varying degrees of detail.
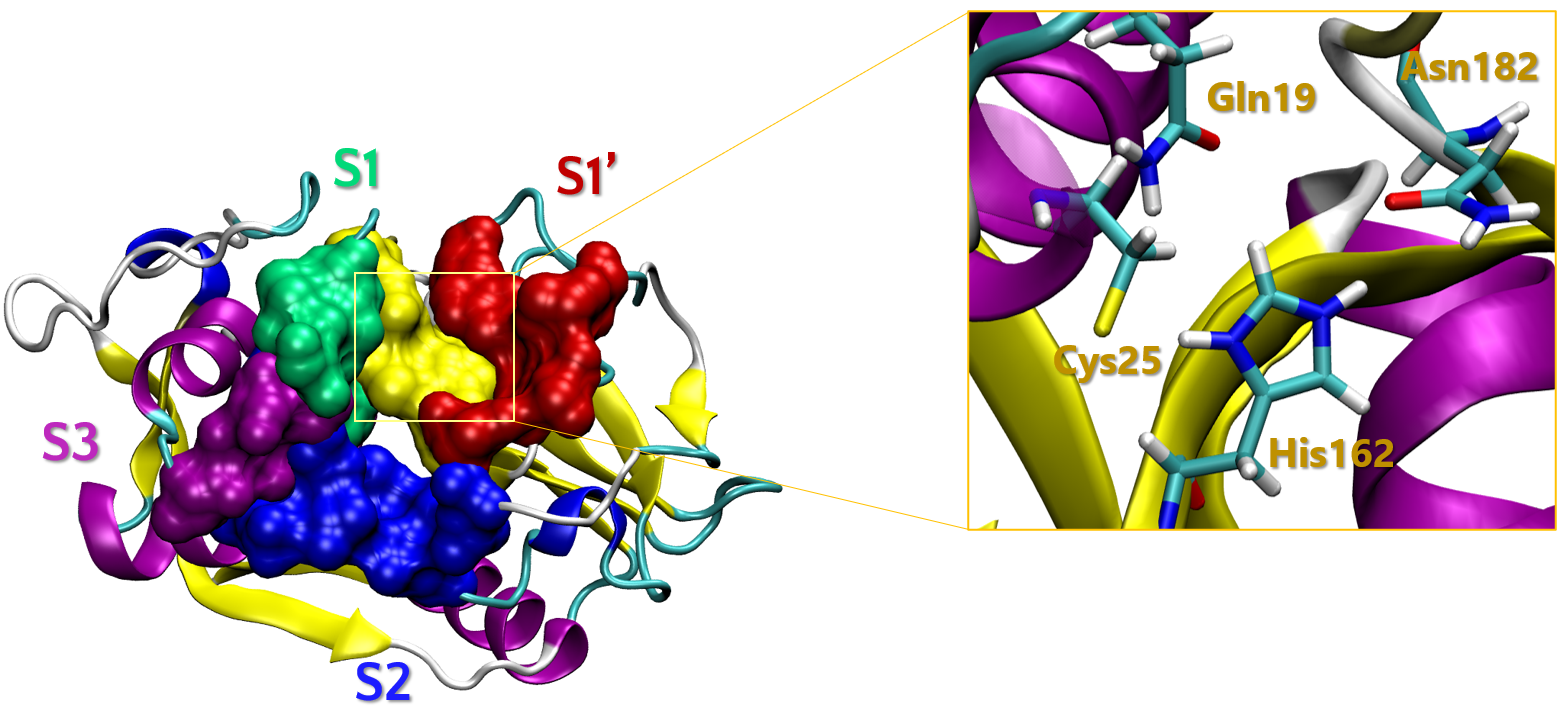
In the publication that we will follow, the researchers aims to perform a combinatorial exloration of R1, R2 and R3 on a central scaffold of a 1,2,3-triazole ring. The core synthetic strategy involves multiple reaction steps, converging into a final step involving a Cu(I)-catalyzed 1,3-dipolar cycloaddition (CuAAC) reaction to obtained the corresponding 1,4-disubsituted 1,2,3-triazoles (Figure 2).
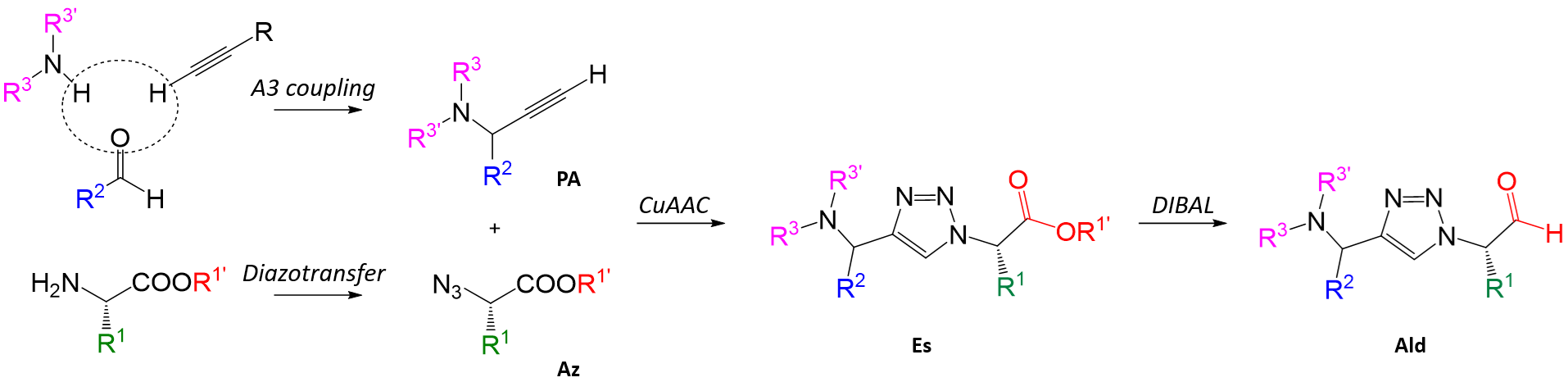
As can be seen, three starting building blocks are required to construct the target chemical space:
- Aminoacids: will provide R1 substituents
- Aldehides: : will provide R2 substituents
- Aliphatic mines: can be either primary or secondary and will provide R3 substituents
The objective of this tutorial is to show how TidyScreen can be used efficiently construct a chemical space of thousands of analogues starting from the corresponding building blocks.
Step 1: Obtention of building blocks
The obtention of the building blocks can be perfomed from different sources. Frequently, a list of readily available reactants in the laboratory constitutes a good source. Alternatively, if large screening campaigns are envisioned, it may be useful to start from commercially available reactants deposited in vendors/public databases.
In this example, we used the eMolecules database as a starting point, which at the moment of writting this tutorial included more than 18 million available reactants.
As a first step, lets create and activate a dedicated TidyScreen project to store, process, filter and synthesize the target library of compounds:
> # Import required modules
>>> from tidyscreen import tidyscreen as ts
>>> from tidyscreen.chemspace import chemspace as chemspace
> # Create a dedicated project for the synthesis
>>> ts.create_project("$HOME/Desktop/example", "synthesis_example")
> # Activate the project just created
>>> synthesis_example = ts.ActivateProject("synthesis_example")
> # Activate the ChemSpace section of the project
>>> synthesis_example_chemspace = chemspace.ChemSpace(synthesis_example)
Next, import the whole eMolecules database into the project database.
> # Input the eMolecules database into ChemSpace from the corresponding 'emolecules.csv' file"
>>> synthesis_example_chemspace.input_csv("$PATH/TO/emolecules.csv")
> # Note_1: we are not providing the full archive corresponding to the emolecules database. The user can obtain it upon request to the authors.
> # Note_2: the input of the whole eMolecules database may take a while to load, since it contains millions of molecules.
Upon reading the reactants, a table named emolecules in the chemspace.db database located within the $PROJECT_PATH/chemspace/processed_data/ folder. A quick snapshot of the generated table is shown in Figure 3:
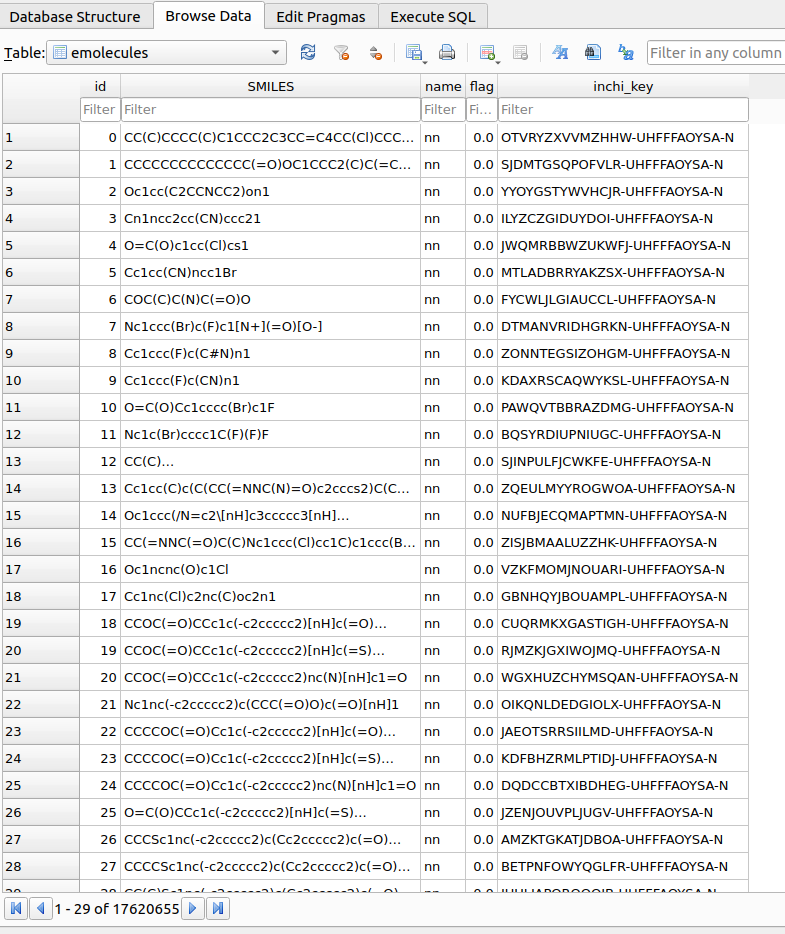
Once the reactants emolecules table has been created, SMARTS filtering procedures can be applied to select the required building blocks. As a first step, lest check the available SMARTS filters. In this case, built-in filters provided upon TidyScreen installation are shown:
> # List available SMARTS filters to obtain reactants
>>> synthesis_example_chemspace.list_available_smarts_filters()
> # Outputs:
>>> Available SMARTS filters:
>>> Filter_id: 1, Filter_Name: Aminoacids, SMARTS: [NX3H2,NX4+H3][CX4H]([*])[CX3H0](=[OX1])[OX2H,OX1-]
>>> ...
>>> Filter_id: 10, Filter_Name: Aldehydes, SMARTS: [CX3H1](=O)[#6]
>>> Filter_id: 11, Filter_Name: PrimAmines, SMARTS: [NX3;H2;!$(NC=O)]
>>> Filter_id: 12, Filter_Name: SecAmines, SMARTS: [NX3;H1;!$(NC=O)]
...
As can be seen in the output, we are interested in using two of the filter provided with TidyScreen installation:
- Filter
1(aminoacids) - Filter
10(aldehydes)
Respect to the filtering of amines, althought there are custom filters matching primary OR secondary amines installed by default, there is no current filter matching both of them. Consequently, we can add our own custom filter:
> # Add a custom filter indicating the SMARTS, followed by a description of the filter
>>> synthesis_example_chemspace.add_smarts_filter("[NX3;H1,H2;!$(NC=O)]","Primary_and_Secondary_Amines")
When listing again, we can see that the filter has been added to the local TidyScreen installation with id:54:
> # List available SMARTS filters to obtain reactants
>>> synthesis_example_chemspace.list_available_smarts_filters()
> # Outputs:
>>> Available SMARTS filters:
>>> Filter_id: 1, Filter_Name: Aminoacids, SMARTS: [NX3H2,NX4+H3][CX4H]([*])[CX3H0](=[OX1])[OX2H,OX1-]
>>> ...
>>> Filter_id: 10, Filter_Name: Aldehydes, SMARTS: [CX3H1](=O)[#6]
>>> Filter_id: 11, Filter_Name: PrimAmines, SMARTS: [NX3;H2;!$(NC=O)]
>>> Filter_id: 12, Filter_Name: SecAmines, SMARTS: [NX3;H1;!$(NC=O)]
...
>>> Filter_id: 54, Filter_Name: Primary_and_Secondary_Amines, SMARTS: [NX3;H1,H2;!$(NC=O)]
From a synthetic point of view and due to the underlying reaction mechanism, there are also additional requisites related to the building blocks structure that further imposes decoration limitations. For example, when filtering aminoacid building blocks, the following criterias are required:
-
only one aminoacid scaffold present, otherwise the reaction will lead to several by products - Filter specification: (1:1);
-
no additional primary amines of carboxyls in R, otherwise the azidation will lead to the mixture of two azides - Filter specification: (11:1 # limit the amine to the primary amine present in the aminoacid; 26:1 # limit the carboxyl to one corresponding to the aminoacid);
-
no azide moiety originally contained in the aminoacid R group - Filter specification: (4:0);
-
no terminal alkynes present in all building blocks, since they will interfere with the A3 coupling and CuAAC reactions - Filter specification: (5:0);
-
no thiols - Filter specification: (25:0);
-
no esters, due to potential stability issues in the final molecules - Filter specification: (35:0);
-
no amides, same as above - Filter specification: (21:0);
-
no sulphonamides - Filter specification: (53:0);
-
no exotic atoms such as: boron, silicon, selenium, 13C, 2H, 3H, 13C, 15N, etc. - Filter specifications: (2:0), (3:0), (17:0), (6:0), (7:0), (8:0), (9:0);
-
The dictionary matching the whole set of filtering for aminoacids is:
{1:1,11:1,26:1,4:0,5:0,25:0,35:0,21:0,53:0,2:0,17:0,6:0,7:0,8:0,9:0}
Also some restrictions applies to the filtering of aldehydes required for A3 coupling reactions:
-
only one aldehyde group - Filter specification: (10:1);
-
no amines groups - Filter specification: (11:0 # No primary amines; 12:0 # No secondary amines);
-
no caboxylic acids groups - Filter specification: (26:0)
-
no azide moiety originally contained in the aminoacid R group - Filter specification: (4:0);
-
no terminal alkynes present in all building blocks, since they will interfere with the A3 coupling and CuAAC reactions - Filter specification: (5:0);
-
no thiols - Filter specification: (25:0);
-
no esters, due to potential stability issues in the final molecules - Filter specification: (35:0);
-
no amides, same as above - Filter specification: (21:0);
-
no sulphonamides - Filter specification: (53:0);
-
no exotic atoms such as: boron, silicon, selenium, 13C, 2H, 3H, 13C, 15N, etc. - Filter specifications: (2:0), (3:0), (17:0), (6:0), (7:0), (8:0), (9:0);
-
The dictionary matching the whole set of filtering for aldehydes is:
{10:1,11:0,26:0,12:0,4:0,5:0,25:0,35:0,21:0,53:0,2:0,17:0,6:0,7:0,8:0,9:0}
An finally, the primary/secondary amines needs to fulfill the following criteria:
-
only one primary/secondary amine group - Filter specification: (54:1);
-
no aldehydes groups - Filter specification: (10:0);
-
no azide moiety originally contained in the aminoacid R group - Filter specification: (4:0);
-
no terminal alkynes present in all building blocks, since they will interfere with the A3 coupling and CuAAC reactions - Filter specification: (5:0);
-
no thiols - Filter specification: (25:0);
-
no esters, due to potential stability issues in the final molecules - Filter specification: (35:0);
-
no amides, same as above - Filter specification: (21:0);
-
no sulphonamides - Filter specification: (53:0);
-
no exotic atoms such as: boron, silicon, selenium, 13C, 2H, 3H, 13C, 15N, etc. - Filter specifications: (2:0), (3:0), (17:0), (6:0), (7:0), (8:0), (9:0);
-
The dictionary matching the whole set of filtering for primary/secondary amines is:
{54:1,10:0,4:0,5:0,25:0,35:0,21:0,53:0,2:0,17:0,6:0,7:0,8:0,9:0}
The set of building blocks restrictions can be further modified/optimized/extended upon discussion with the wet-lab team in charge of performing the synthetic procedures, further adapting the requirements to the empirical experimental versatility.
Step 2: Construction and execution of a filtering workflow
It is possible to concatenate multiple filters in a single filtering workflow so as to comply with all the above-mentioned criteria at once. For example:
> # Create a workflow to filter aldehydes (10:1) for A3 coupling reactions (no interfering groups as indicated: 12:0, 11:0, etc)
>>> synthesis_example_chemspace.create_smarts_filters_workflow({10:1,11:0,26:0,12:0,4:0,5:0,25:0,35:0,21:0,53:0,2:0,17:0,6:0,7:0,8:0,9:0})
> # Create a workflow to filter primary and secondary amines (54:1) for A3 coupling reactions reactions
>>> synthesis_example_chemspace.create_smarts_filters_workflow({54:1,10:0,4:0,5:0,25:0,35:0,21:0,53:0,2:0,17:0,6:0,7:0,8:0,9:0})
> # Create a workflow to filter aminoacids (1:1) for click reactions
>>> synthesis_example_chemspace.create_smarts_filters_workflow({1:1,11:1,26:1,4:0,5:0,25:0,35:0,21:0,53:0,2:0,17:0,6:0,7:0,8:0,9:0})
Once created, available reactants filtering workflows can be listed:
> # List available filtering workflows
>>> synthesis_example_chemspace.list_available_smarts_filters_workflows()
> ### Outputs
>>> Available SMARTS filters workflows:
> # Workflow to filter aldehydes
>>> Workflow_id: 1, Filter_Specs: {"10": 1, "11": 0, "26": 0, "12": 0, "4": 0, "5": 0, "25": 0, "35": 0, "21": 0,"53": 0, "2": 0, "17": 0,"6": 0, "7": 0, "8": 0,"9": 0}, Description: Filter Aldehydes to perform A3 coupling reactions
> # Workflow to filter primary/secondary amines
>>> Workflow_id: 2, Filter_Specs: {"54": 1, "10": 0,"4": 0, "5": 0, "25": 0, "35": 0, "21": 0, "53": 0,"2": 0, "17": 0, "6": 0, "7": 0, "8": 0, "9": 0}, Description: Filter primary amines for A3 coupling reactions
> # Workflow to filter aminoacids
>>> Workflow_id: 3, Filter_Specs: {"1": 1, "11": 1, "26": 1, "4": 0, "5": 0, "25": 0, "35": 0, "21": 0,"53": 0, "2": 0, "17": 0, "6": 0, "7": 0, "8": 0, "9": 0}, Description: Filter Aminoacids for Click reactions
A filtering workflow can be applied on a given table (i.e. emolecules) that has already been stored in the chemspace.db using:
> # Apply workflow_filter_id: 1 on the 'emolecules' table
>>> synthesis_example_chemspace.subset_table_by_smarts_workflow("emolecules",1)
Upon executing the filter, a table named tables_subsets will be created in chemspace.db with the objective of registering the filtering action:
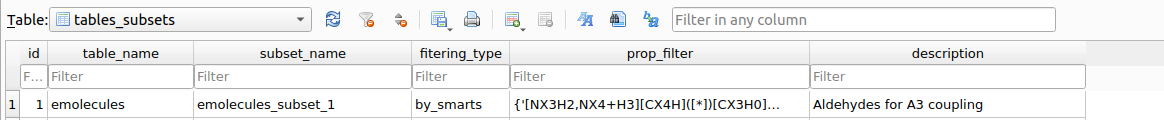
tables_subsets containing the filtering actions.The info included in the tables_subsets is:
table_name: the source table on which the filtering workflow was applied.subset_name: the destination table to which the filtered compounds were written. This name is automatically created incrementally.filtering_type: the kind of filtering that originated the destination table. In this caseemolecules_subset_1was created by using SMARTS filtering (filteringby_propertiesis also possible, as we will see later)prop_filter: explicit indication ot the filtering workflow applied.description: this is requested to the used as descriptive information upon executing the filtering workflow.
After filtering the required aldehydes, amines and aminoacids, tables_subsets looks like this:
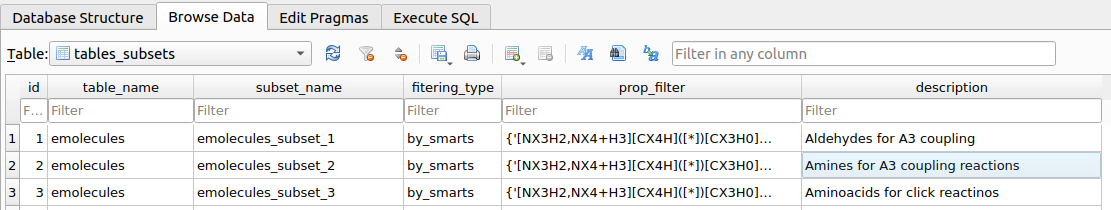
tables_subsets after filtering aldehydes, amines and aminoacids.In this specific example, the resulting filtered tables contained:
- Aldehydes: 227,985 compounds
- Primary/Secondary amines: 1,420,273
- Aminoacids: 5,405
A random set of each subseted table can be depicted:
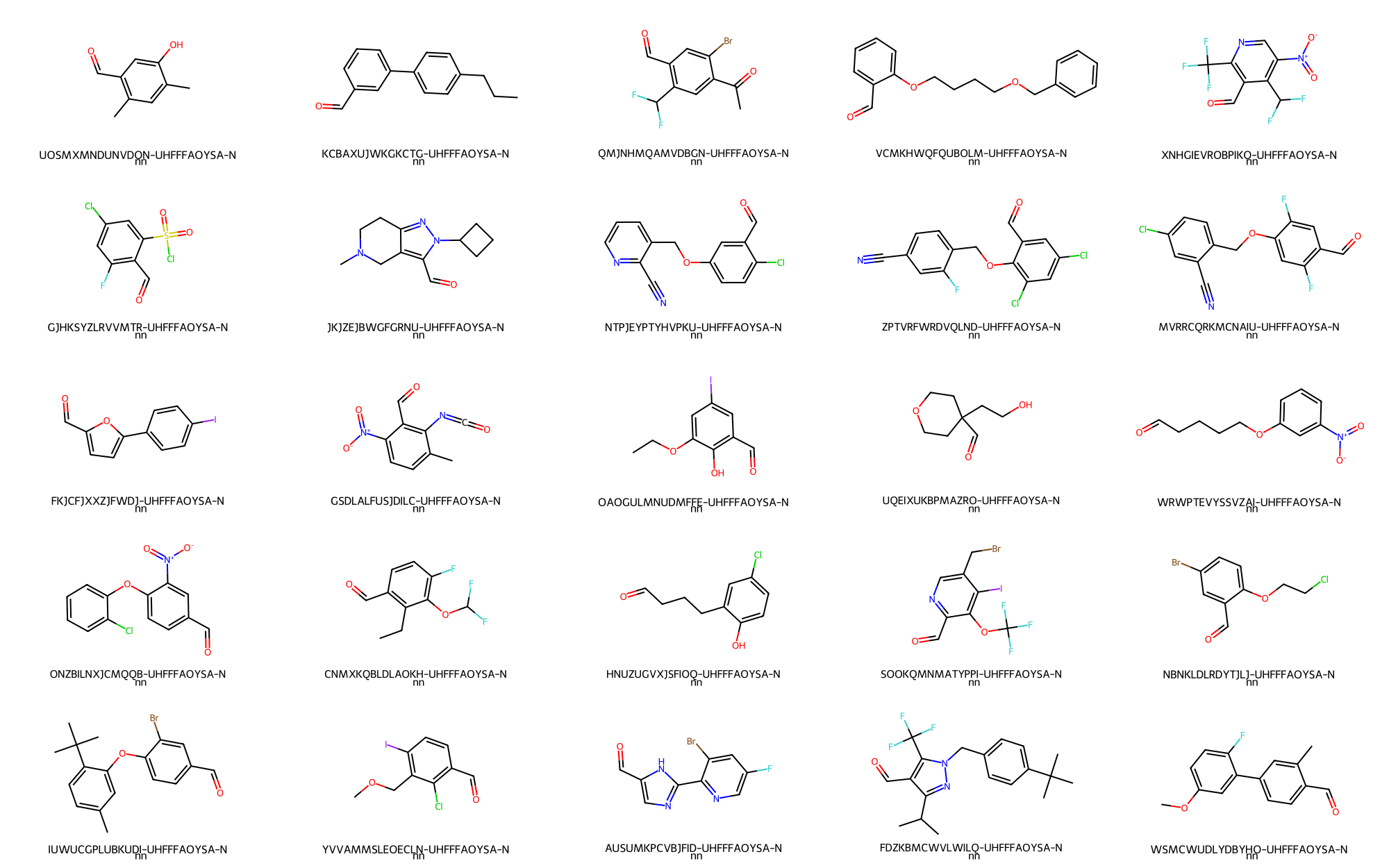
> # Randomly depict 25 aldehydes
>>> synthesis_example_chemspace.depict_ligand_table("emolecules_subset_1", limit=25,random=True)
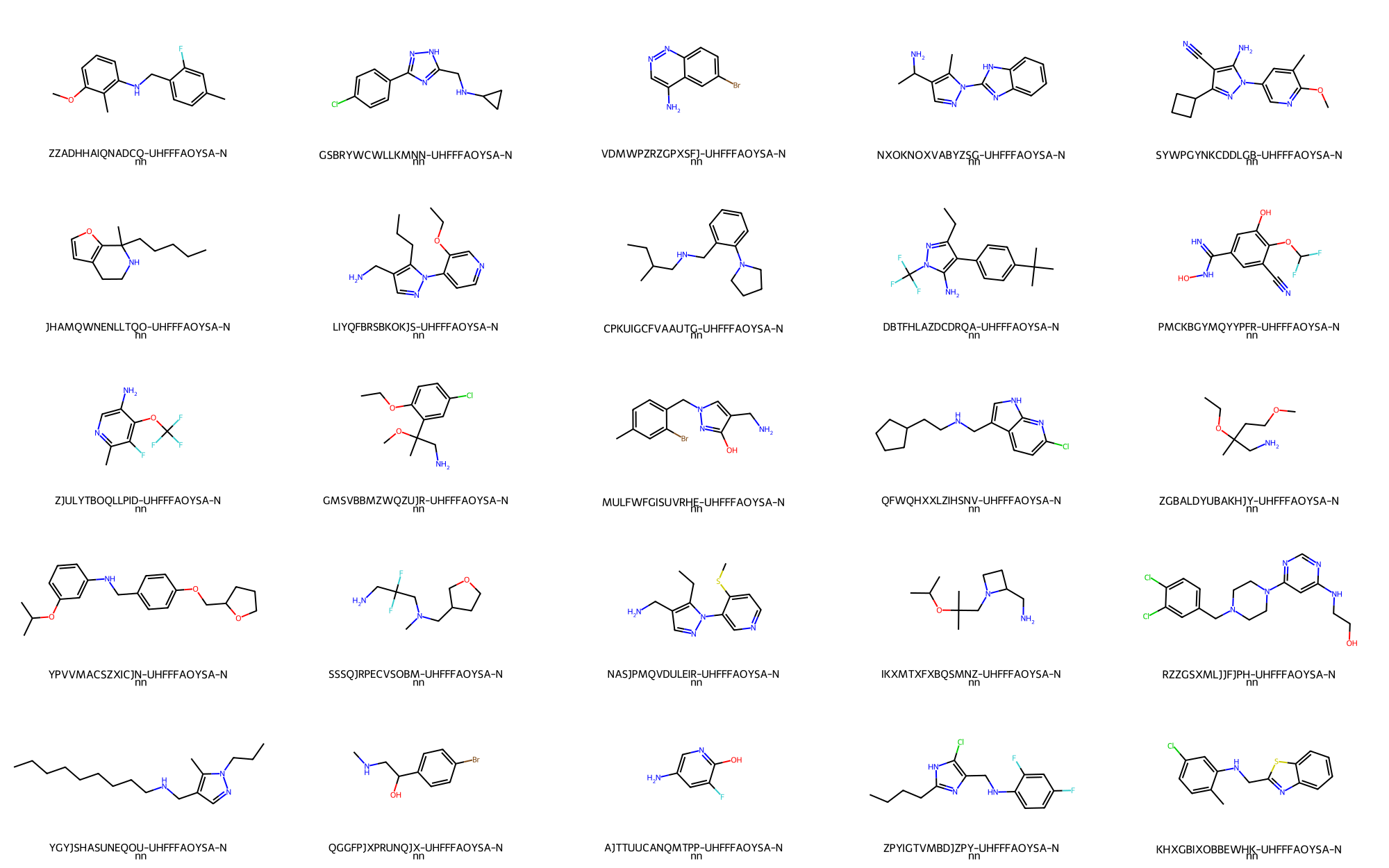
> # Randomly depict 25 primary amines
>>> synthesis_example_chemspace.depict_ligand_table("emolecules_subset_2", limit=25,random=True)
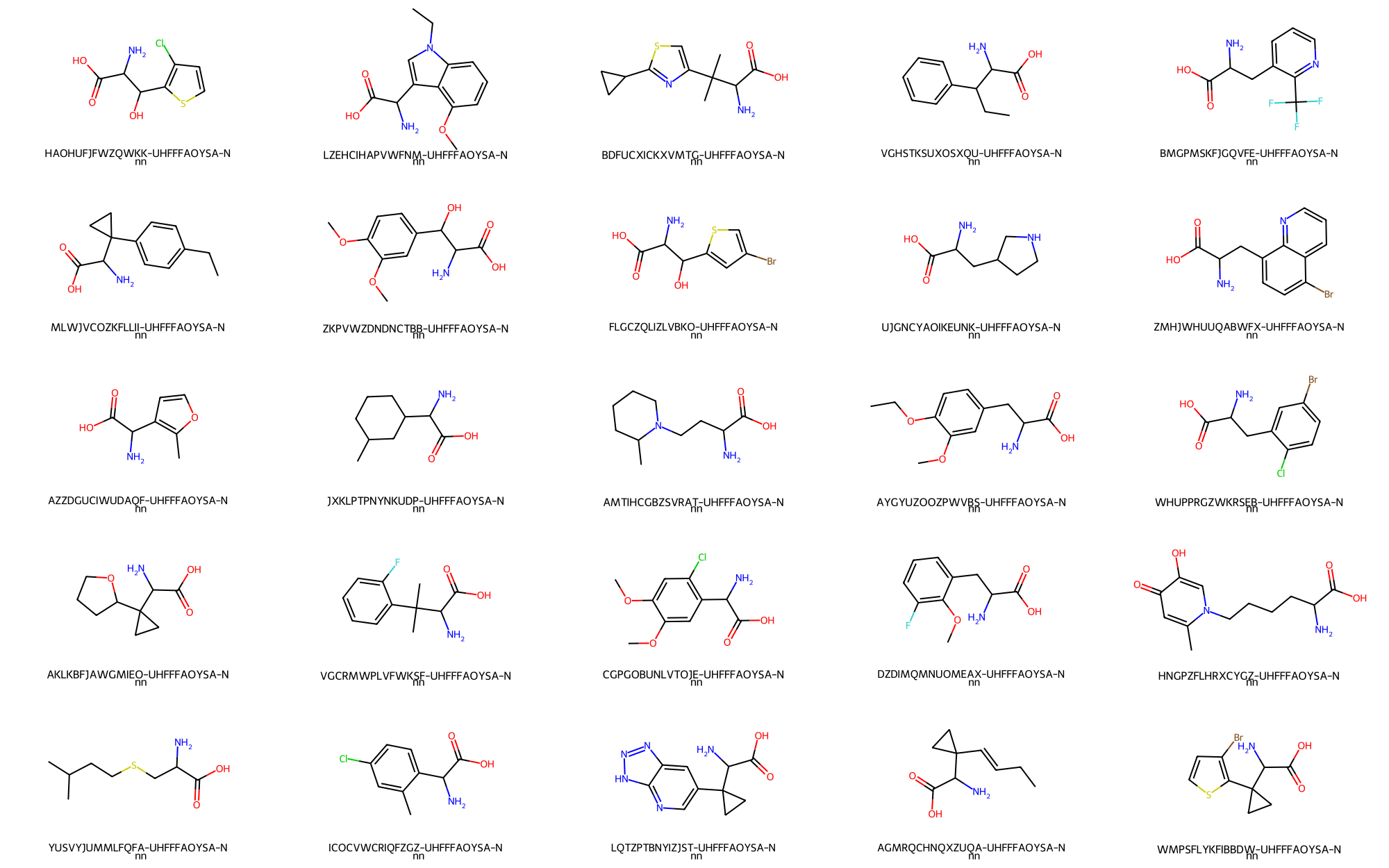
> # Randomly depict 25 aminoacids
>>> synthesis_example_chemspace.depict_ligand_table("emolecules_subset_3", limit=25,random=True)
The resulting depictions are:
- Aldehydes -> 25 / 227,985
- Primary/Secondary amines -> 25 / 1,420,273
- Aminoacids -> 25 / 5,405
It is clear that exploring the while set of combinatorial possibilities towards 1,4 disubstituted 1,2,3-triazoles is not possible in terms of the enormous number of compounds that would be generated (n = 227,985 * 1,420,273 * 5,405). Thus, some filtering/prioritization criteria, such as by drug-like properties, may be accomplished on the resulting subsets. In this way, different physicochemical properties can be computed for each table subset as follows:
> ## By default the properties are computed: ["MolWt","MolLogP","NumHDonors","NumHAcceptors","NumRotatableBonds","TPSA"]
> ### You can specify whatever property computed by RDKit you wan using the 'properties_list' keyword
>>> synthesis_example_chemspace.compute_properties("emolecules_subset_1")
After computing the properties, the emolecules_subset_1 contains additional columns including de calculated values:
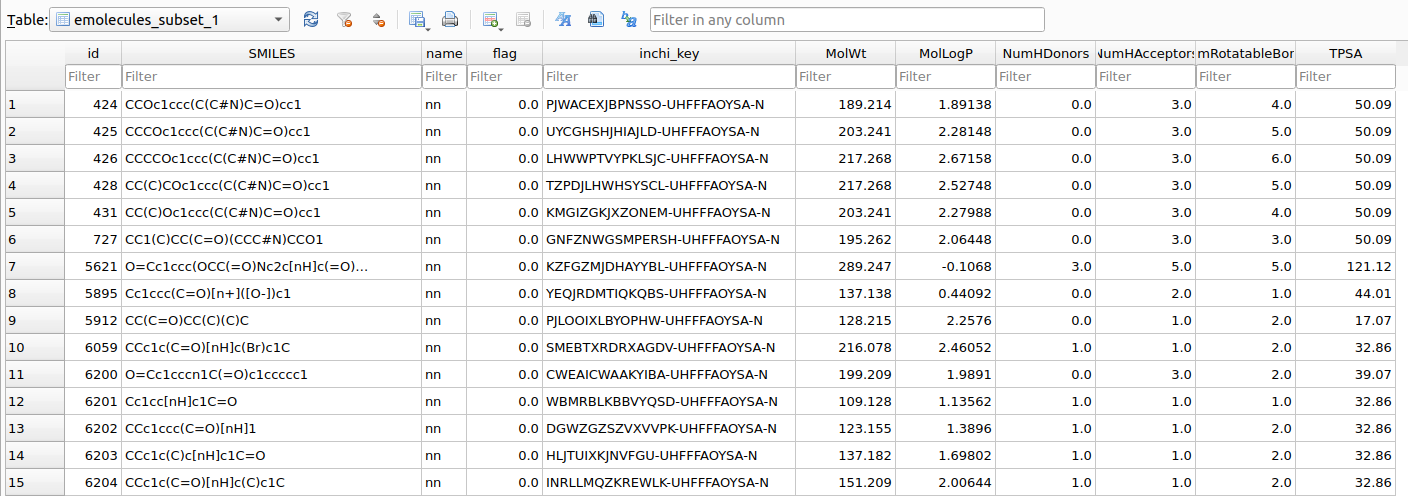
emolecules_subset_1 after computing default physicochemical properties.It is now possible to subset a given table containing computed properties. As an example, lets use the following criteria:
MolWt>= 200 andMolWt<= 500MolLogP>=1.5 andMolLogP<= 3NumRotatableBonds<= 2
In order to combine the filtering criteria, it is possible to construct a Python list indicating the threshold values as follows: ["MolWt>=200","MolWt<=500","MolLogP>=1.5","MolLogP<=3","NumRotatableBonds<=2"]. This list can now be passed the corresponding subsetting method in TidyScreen:
> # Subset 'emolecules_subset_1' using the given properties thresholds
>>> synthesis_example_chemspace.subset_table_by_properties("emolecules_subset_1",["MolWt>=200","MolWt<=500","MolLogP>=1.5","MolLogP<=3","NumRotatableBonds<=2"])
After executing the filtering, a record of the action will be stored in the tables_subsets table:
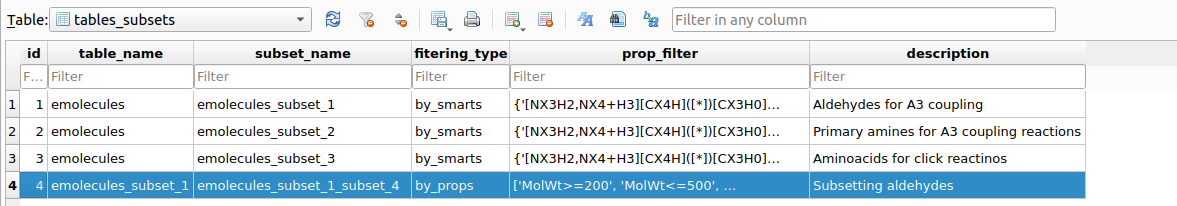
tables_subsetswhich registers the filtering/subseting actions.As can be seen the table emolecules_subset_1_subset_4 has been created based on the corresponding filtering workflow. It is advisable to retain this table name for tracebility of the reactants filterings history. If the user wants to copy this subsetted table with a more comprehensive name, it can be done as follows:
> ## Copy the subseted table with a new name
>>> synthesis_example_chemspace.copy_table_to_new_name("emolecules_subset_1_subset_4","aldehydes_for_A3_coupling")
In case the user wants to save the filtered table as a .csv file for storing purposes use:
> # Save a .csv file with the filtered table
>>> synthesis_example_chemspace.save_table_to_csv("emolecules_subset_1_subset_4")
For the purposes of this example and following the procedures applied by Cerutti et al, custom drug-like filtering criterias were used to reproduce the bulding blocks selection, leading to a set of:
- Aldehydes : 51 building blocks
- Primary/Secondary amines : 22 building blocks
- Aminoacids : 35 building blocks
The combinatorial synthesis of the building blocks will lead to a library of 39,270 1,2,3-triazoles.
Definition of single SMARTS reactions
It is now the moment to create the corresponding single reactions to be combined into the overall synthetic workflow. It should be noted that SMARTS based reactions that are created by the user belongs only to this specific project (i.e. they are not stored system-wide as chemical filters are). The reason behind this implementation is that these reactions are dependent on the specific synthetic plan associated to the project.
In order to list available reactions for the current project, use the corresponding method:
> # List available reactions in the current project
>>> synthesis_example_chemspace.list_available_smarts_reactions()
> # Outputs
>>> "SMARTS reactions table does not exist yet. Add reactions to the database first."
Note that the first time you run this listing, since no reactions are available within the project, the message "SMARTS reactions table does not exist yet. Add reactions to the database first." will be printed to the terminal. To add user defined reactions in the SMARTS formats proceed as follows:
> # Template to add single reactions to the project database
>>> synthesis_example_chemspace.add_smarts_reaction("SMARTS_REACTION", "Description")
In the context of the synthetic plan shown in Figure 2, we are interested in chaining the following reactions as part of our synthetic scheme:
- Diazotransfer on aminocids:
[NX3;H2:1][CX4:2][CX3,H0:3]>>[N-]=[N+]=[NX2;H0:1][CX4:2][CX3,H0:3] - Acylation of carboxylic acids:
[CX3:1](=[O:2])[OX2H,OX1-:3].[C:4][O]>>[C:1](=[O:2])[O:3][C:4] - A3 Coupling reaction:
[N:1].[CX3H1:2](=[O:3])>>[N:1][C:2][C:4]#[C:5] - CuAAC reaction:
[NX1-:1]=[NX2+:2]=[NX2:3].[CX2H1:4]#[CX2H0:5]>>[NX2+0:1]1=[NX2+0:2][N:3]-[C:4]=[C:5]1 - DIBAL reduction:
[CX3:1](=[O:2])[OX2H,OX1-:3]>>[CX3H1:1](=[O:2])
The construction of SMARTS based reactions should be carefully analyzed and tested. There are several tutorials in the web on how to construct them. This is an example.
At this point, we should add the corresponding reactions to the project database:
> # Add the diazotransfer reaction
>>> synthesis_example_chemspace.add_smarts_reaction("[NX3;H2:1][CX4:2][CX3,H0:3]>>[N-]=[N+]=[NX2;H0:1][CX4:2][CX3,H0:3]", "Diazotransfer")
> # Add a general acylation reaction
>>> synthesis_example_chemspace.add_smarts_reaction("[CX3:1](=[O:2])[OX2H,OX1-:3].[C:4][O]>>[C:1](=[O:2])[O:3][C:4]", "Acylation")
> # Add the A3 coupling reaction
>>> synthesis_example_chemspace.add_smarts_reaction("[N:1].[CX3H1:2](=[O:3])>>[N:1][C:2][C:4]#[C:5]", "A3 coupling")
> # Add the CuAAC reaction
>>> synthesis_example_chemspace.add_smarts_reaction("[NX1-:1]=[NX2+:2]=[NX2:3].[CX2H1:4]#[CX2H0:5]>>[NX2+0:1]1=[NX2+0:2][N:3]-[C:4]=[C:5]1", "CuAAC")
> #Add the DIBAL reduction reaction
>>> synthesis_example_chemspace.add_smarts_reaction("[CX3:1](=[O:2])[OX2H,OX1-:3]>>[CX3H1:1](=[O:2])", "DIBAL reduction")
Upon adding reactions, a table named smarts_reactions will be created in the chemspace.db database:
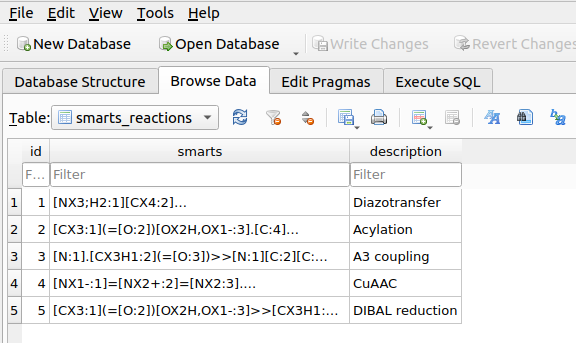
Creation and execution of a reaction workflow
Once reactions are available within the project, they can be executed as single/multiples steps combined into a reaction workflow. Reaction steps can also products generated within the same workflow. In order to check the correctness of the reactions definition, it is a good idea to incrementally construct the final workflow, performing dry_runs to check if the reactants were processed adequately.
Lets start by applying the A3 coupling reaction:
> # Create a workflow involving a single A3 coupling step (reaction id: 3)
>>> synthesis_example_chemspace.add_smarts_reaction_workflow([3])
A new table named smarts_reactions_workflow has been created in chemspace.db:
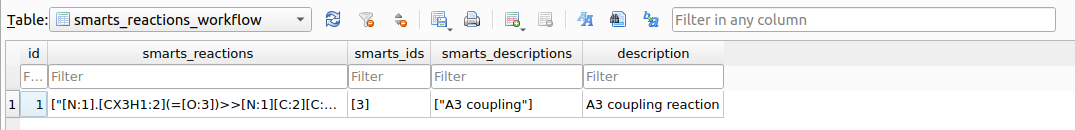
smarts_reactions_workflow table containing available synthetic pipelines.Available reaction workflows can also be printed to the terminal using:
> # List available reaction workflows
>>> synthesis_example_chemspace.list_available_reactions_workflows()
Now lets execute a test (i.e. select a dry_run) using the reaction workflow id '1' and providing the corresponding reactants:
> # Execute the reaction workflow id = 1 (A3 coupling reaction)
>>> synthesis_example_chemspace.apply_reaction_workflow(1,[["Amines_filtered_druglike","Ald_filtered_druglike"]],dry_run=1)
> # Outputs:
>>> Checking the reaction workflow definition vs the reactants lists: OK
>>> apply_single_bimolecular_reaction_step
>>> React.Step 0: - Reaction: [N:1].[CX3H1:2](=[O:3])>>[N:1][C:2][C:4]\#[C:5] - Reactants1: 22 - Reactants2: 51 - Products: 1122
As can be seen, the system validation of the workflow definition passed ok, indicating that a single bimolecular reaction step was detected, further informing and applying the reaction SMARTS. Combination of 22 amines with 51 aldehydes should originate 1122 product, which is in aggreement to what is informed. Consequently this reaction step is validated.
Lets add and execute the azide formation step starting from aminoacid building blocks:
> # Create a workflow involving a single diazotransfer step (reaction id: 1)
>>> synthesis_example_chemspace.add_smarts_reaction_workflow([1])
> # Execute reaction workflow id = 2 (diazotransfer reaction)
>>> synthesis_example_chemspace.apply_reaction_workflow(2,[["AA_filtered_druglike"]],dry_run=1)
> # Outputs:
>>> Reaction step 0 applied successfully. Products stored in column: 'SMILES_product_step_0'
>>> Reaction step 0: - Reaction: [NX3;H2:1][CX4:2][CX3,H0:3]>>[N-]=[N+]=[NX2;H0:1][CX4:2][CX3,H0:3] - Number of reactants: 35 - Number of products: 35
Again the number of products is consistent with which was expected, confirming that the reaction has been adequately defined.
Let combine and execute all the steps involved in the synthetic scheme (A3 coupling -> azidotransfer -> CuAAC) into a multistep workflow :
> # Create a workflow involving a single diazotransfer step (reaction id: 1)
>>> synthesis_example_chemspace.add_smarts_reaction_workflow([3,1,4])
> # Execute reaction workflow id = 3 (chained synthetic scheme)
>>> synthesis_example_chemspace.apply_reaction_workflow(3,[["Amines_filtered_druglike","Ald_filtered_druglike"],["AA_filtered_druglike"],["->:-1","->:-2"]],dry_run=1)
> # Outputs
>>> Checking the reaction workflow definition vs the reactants lists: OK
>>> apply_single_bimolecular_reaction_step
>>> React.Step 0: - Reaction: [N:1].[CX3H1:2](=[O:3])>>[N:1][C:2][C:4]\#[C:5] - Reactants1: 22 - > Reactants2: 51 - Products: 1122
>>>
>>> Reaction step 1 applied successfully. Products stored in column: 'SMILES_product_step_1'
>>> Reaction step 1: - Reaction: [NX3;H2:1][CX4:2][CX3,H0:3]>>[N-]=[N+]=[NX2;H0:1][CX4:2][CX3,H0:3] - > Number of reactants: 35 - Number of products: 35
>>>
>>> Previous step references: 1, 2
>>> apply_single_bimolecular_reaction_step
>>> React.Step 2: - Reaction: [NX1-:1]=[NX2+:2]=[NX2:3].[CX2H1:4]\#[CX2H0:5]>>[NX2+0:1]1=[NX2+0:2][N:3]->[C:4]=[C:5]1 - Reactants1: 35 - Reactants2: 1122 - Products: 39270
Note how the reactants for the third reaction step (CuAAC) have been defined:
"->:-1": means "input the products of the previous step (-1, Azides)""->:-2": means "input the products of the two reaction steps before (-2, alkynes obtained from the A3 coupling reaction)"
As can be seen, the combinatorial number of triazoles generated (n = 39270) is in agreement with what we expected.
With the definition of the reaction workflow being checked, we can now run it in production mode:
> # Create a workflow involving a single diazotransfer step (reaction id: 1)
>>> synthesis_example_chemspace.add_smarts_reaction_workflow([3,1,4])
> # Execute reaction workflow id = 3 (chained synthetic scheme)
>>> synthesis_example_chemspace.apply_reaction_workflow(3,[["Amines_filtered_druglike","Ald_filtered_druglike"],["AA_filtered_druglike"],["->:-1","->:-2"]])
After executing the synthetic workflow, a registry is generated in the table reactions_attempts within the chemspace.db database:
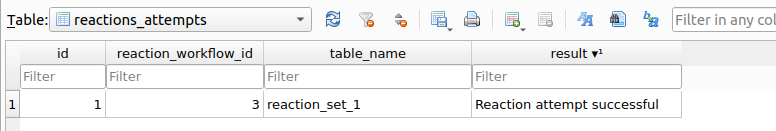
reactions_attempts table indicating the execution of the synthetic workflow.As shown, the table reaction_set_1 containing the reaction products has been created. Lets depict a subset of 25 products stored within it:
> # Randomly depict 25 1,2,3-triazoles obtained by combinatorial virtual synthesis
>>> synthesis_example_chemspace.depict_ligand_table("reaction_set_1", limit=25,random=True)
- A set of 25 1,4 disubstituted 1,2,3-triazoles generated by virtual synthesis_
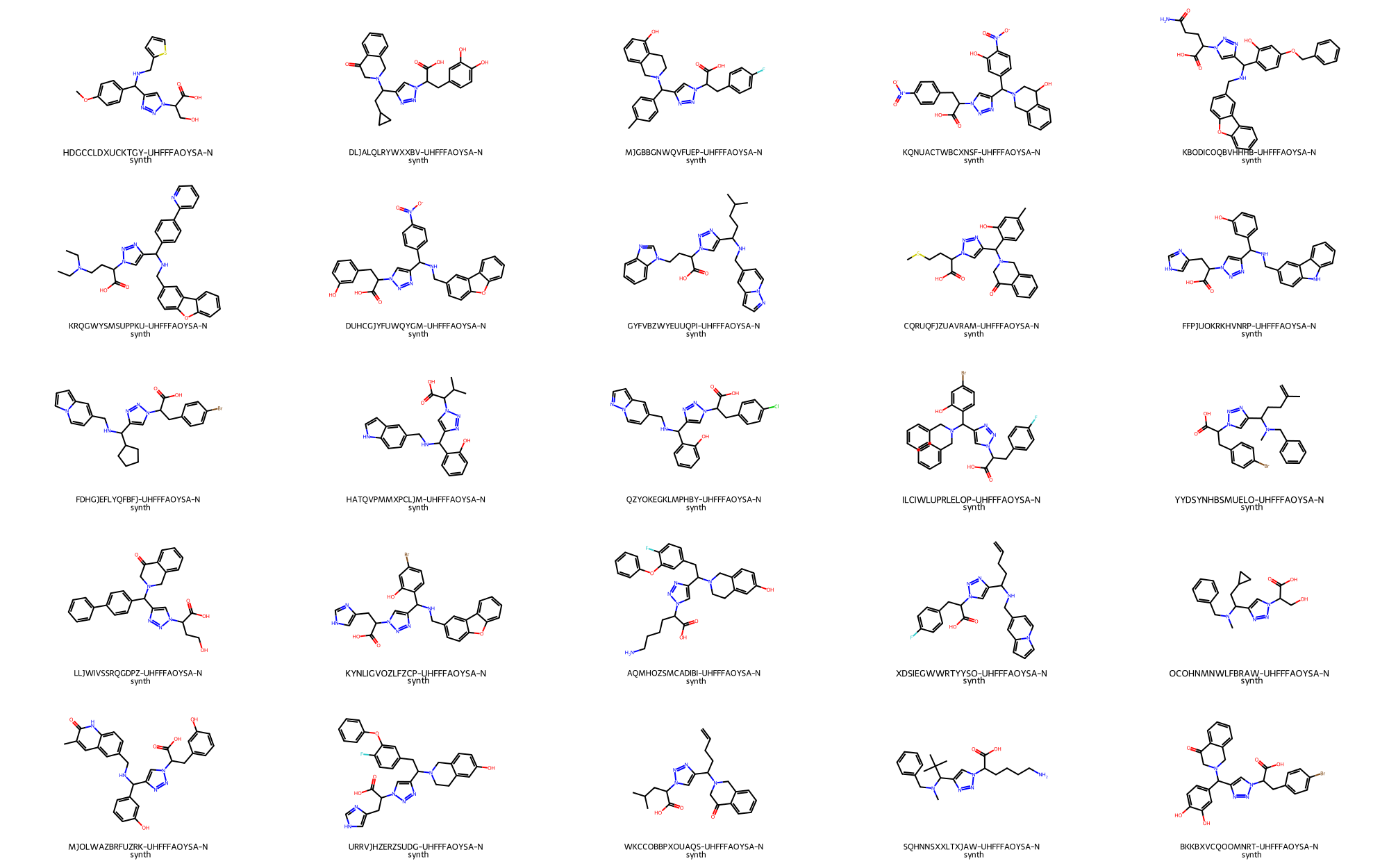
At this point, library of 1,4 disubstituted 1,2,3-triazoles contained within table reaction_set_1 is ready for further processing towards the screening of potential CZP targeted covalent inhibitors. Some of the steps following the virtual screening campaing may encompass:
- Computation of drug-like properties and chemical space sampling/prioritization of the 39,270 triazoles synthesized virtually;
- Further chemical modifications by applying new synthetic workflows, such as warhead modifications on this set of 39,270 triazoles;
- Preparation and execution of molecular docking docking studies on a CZP receptor model/s.
- Etc.
Just play with TidyScreen and be creative!
"Creativity is intelligence having fun." — Albert Einstein