Running a TidyScreen project
A typical vHTS project is comprised by three main stages:
- Construction of a working chemical space;
- Molecular docking of selected candidates;
- Molecular dynamics simulatons on promising hits;
TidyScreen assists in the configuration and execution of each of these stages. The central strategy is based on the philosophy that indexing and storing data in SQL databases can be functional not only to the organization of each working stage, but also favors the natural interconnection between them.
Construction of a working chemical space
The chemical space to be explored during a vHTS campaign can be constructed using diverse methodological approaches, each one particularly adapted to the research objectives. Some of the alternatives are:
- Reading compounds from a custom file;
- Filtering of commercially available compounds;
- Construction of libraries from databases (such as ChEMBL);
- Exploration of in-house compounds libraries;
- Enumeration of synthetic chemical routes;
- Etc...
Reading compounds from a custom file
This is the most simple way of introducing one or more compounds into a TidyScreen project. As mentioned before, these compounds will be stored in the SQL database corresponding to the chemspace dimension. Note: When a file containing compounds is uploaded into TidyScreen, a copy of such file is stored in the 'chemspace/raw' folder.
This is a sample .csv file containing five aliphatic alcohols, and we can use it as and example to store the corresponding compounds into the chemspace database using the following script:
...
## We will add compounds present in 'aliphatic_alcohols.csv' into the already existing 'test1' project
# Activate a project to perform actions
test1_proj = proj_mg.ActivateProject("test1")
# Once activated, initialize ChemSpace actions on the project
test1_proj_CS = proj_mg.ChemSpaceActions(test1_proj)
# Once the ChemSpace dimension is activated, load the compounds present as SMILES in the .csv file
test1_proj_CS.process_raw_cpds_file("$PATH_TO_FILE/aliphatic_alcohols.csv")
Different parsing options can be applied to the .csv file depending on the separator used, the existence of a header or compounds names. In order to let TidyScreen know how to process the file, when executing the script the first two lines will be printed to the terminal and serie of questions will be presented in order to guide the parsing process. The following responses should be provided:
Header presence (y/n): # in this sample file it indeed contains a header (SMILES,Name)
Separator string: # In this case ','
Field number corresponding to SMILES: # in this file '0'
Presence of molecules names (y/n): # in this case 'y'
Field number corresponding to the Names: # in this case '1'
Upon providing the requested information, all molecules corresponding to the SMILES present in the .csv file will be stored in a SQL table named after the provided file (i.e. 'aliphatic_alcohols') and saved into the '/processed_data/chemspace.db' database file of the chemspace domain. (See the
Projects folders structure
section)
Once loaded, the table containing the compounds can be inspected or modified using different approaches, such as:
- Listing the tables present in '/processed_data/chemspace.db' database file of the
chemspacedomain:
...
test1_proj = proj_mg.ActivateProject("test1")
test1_proj_CS = proj_mg.ChemSpaceActions(test1_proj)
# Execute the listing method on the project ChemSpace object
test1_proj_CS.list_ligands_tables()
output:
Tables in database corresponding to project: 'test1':
aliphatic_alcohols # <- the table containing the loaded compounds
# At the moment only one table of compounds is present in the database.
# If more tables are loaded, they will appear in alpahbetical order in the output.
- Listing of compounds present in a given table:
...
test1_proj = proj_mg.ActivateProject("test1")
test1_proj_CS = proj_mg.ChemSpaceActions(test1_proj)
# List the compounds present in the 'aliphatic_alcohols' table.
test1_proj_CS.list_cpds_in_table("aliphatic_alcohols")
output
('CO', 'methanol')
('CCO', 'ethanol')
('CCCO', 'propanol')
('CCCCO', 'butanol')
('CCCCCO', 'pentanol')
A total of 5 molecules are present in table: 'aliphatic_alcohols'
-
Using dedicated tools to manage SQL databases:
-
Use VISIDATA to explore
chemspace.dbin the terminal. -
Use DB Browser to explore
chemspace.dbusing a GUI.
-
-
Deleting a loaded table:
...
test1_proj = proj_mg.ActivateProject("test1")
test1_proj_CS = proj_mg.ChemSpaceActions(test1_proj)
# The 'aliphatic_alcohols' can be removed from 'chemspace.db'.
test1_proj_CS.delete_cpds_table("aliphatic_alcohols")
After a table containing molecules represented in the SMILES format is loaded into TidyScreen, it is sometimes useful to visually inspect them by depicting the structures. This can be done as follows:
t1_proj = proj_mg.ActivateProject("test1")
test1_proj_CS = proj_mg.ChemSpaceActions(test1_proj)
# Depict compounds present in 'aliphatic_alcohols' table. One or more .pn files will be written to the /chemspace/misc/$TABLE_NAME' directory.
test1_proj_CS.plot_table_of_molecules("aliphatic_alcohols")
The file named 'aliphatic_alcohols_0.png' will be written to /chemspace/misc/$TABLE_NAME.
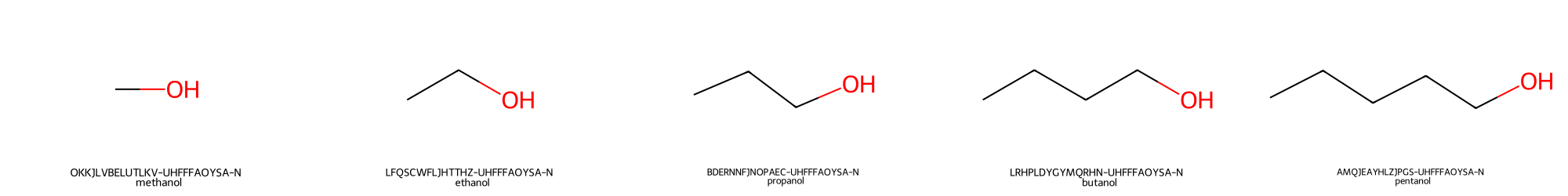
Consider that if a table containing a large number of compounds is depicted, several .png files will be generated, each one containing 25 (5x5) depicted molecules. Each molecule depiction is accompanied by two labels:
-
inchi_key: that is a unique identifier of the molecule, which is automatically computed by TidyScreen at the moment of reading the input file. This identifier will be used as the unique id of the molecule during the vHTS campaign.
-
mol_name: corresponding the the name that was provided by the user un the input file.
Filtering of commercially available compounds;
Considering that most databases containing commercially available compounds are obtained using .csv file, their loading into TidyScreen follows the same procedure described in the previous section. However, since these databases typically contains millions of molecules, it is frequently required to subset the whole database in order to isolate specific type of molecules. TidyScreen provides methods to apply custom chemical filter based on SMARTS notation.
Two frequently used databases of commercially available compounds are ZINC or Emolecules. In this link you will find a .csv file containing a sample (50K molecules) of the Emolecules database in order to test TidyScreen functioning.
Here we load the sample file into the corresponding chemspace table:
...
# Activate 'test1' project
test1_proj = proj_mg.ActivateProject("test1")
# Initialize ChemSpace actions on 'test1'
test1_proj_CS = proj_mg.ChemSpaceActions(test1_proj)
# Load the raw file 'emolecules_sample.csv'
test1_proj_CS.process_raw_cpds_file("$PATH_TO_FILE/emolecules_sample.csv")
After executing the script, TidyScreen will use available cpu cores to load the 50K structures into the corresponding table.
Now, we can subset the newly created 'emolecules_sample' table. This filtering can be performed by using SMARTS notation, and also TidyScreen provides a set of built-in custom filters used to match typical reactants. The list of available filters can be que queried as follows:
from tidyscreen import config as ts_cfg
import tidyscreen_dbs
main_database = ts_cfg.MainDbConfigs()
main_database.list_available_chem_filter()
which will output the following:
1: Aminoacids - [NX3H2,NX4+H3][CX4H]([*])[CX3H0](=[OX1])[OX2H,OX1-]
2: Boron - B
3: Silicon - [Si]
4: Azide - N=[N+]=[N-]
5: Terminal_alkyne - C#[CH]
6: Deuterium - [2H]
...
As can be seen, each chemical filter is identified by a filter_id followed by a filter_nameand filter_smarts. The use of the filter in production is performed by spcifing the corresponding filter_id which is unique to each filter. During the isolation of building blocks that are or interest in a vHTS campaign it is frequently required to build a filters pipeline, i.e. a set of filters that are applied sequently to match chemical conditions. In this way, TidyScreen filters molecules based on these kind of filters pipeline, which can contain one or multiple filters.
A single filter pipeline can be constructed as follows:
...
# Activate 'test1' project
test1_proj = proj_mg.ActivateProject("test1")
# Initialize ChemSpace actions on 'test1'
test1_proj_CS = proj_mg.ChemSpaceActions(test1_proj)
# Create a single filter including id: 1 (Aminoacids)
test1_proj_CS.create_filters_pipeline()
When this script is executed, the following information will be requested:
- Filter number to add: corresponds to the
idof the filter stored in the main database. If entering a multifilter pipeline, new filters will be recursively requested, so end the pipeline the word 'quit' should be entered. In this example we intend to filter aminoacids, so we will indicate '1'. - Number of instances allowed: set the maximum number of filter matches to allow. Think that a given chemical moiety can be present more than once in a molecule, and we may want to filter molecules containing lets stay 'no more than two hydroxyl' groups. In this case we will indicate '1' (i.e. only 1 aminoacid scaffold should be retained).
- Indicate inclusion/exclusion: if inclusion is selected, when the chemical filter is matched the molecule will be retained in the results table. On the other hand, when exlusion is indicated, it means that if matched, the molecule will be discarded from from the results table. In this case we indicate 'I'.
- Pipeline name: finally, when the pipeline has been defined, a 'pipeline name' is requested in order to identify it (we will give this pipeline the name 'aminoacids').
Once created, we can list the filters pipeline that are available for a given project. Please note that filters pipelines are specific to project, while the individual filter database is common to the local TidyScreen installation.
Note: add the script to show the filters pipeline available in the project
At this point we can apply the newly created filter as follows:
...
# Activate 'test1' project
test1_proj = proj_mg.ActivateProject("test1")
# Initialize ChemSpace actions on 'test1'
test1_proj_CS = proj_mg.ChemSpaceActions(test1_proj)
# The 'filters_pipeline' can be applied by indicanting the corresponding `filter_id` and `table_name`
test1_proj_CS.apply_filters_pipeline(1,"emolecules_sample")
If everything works ok, 7 molecules containing filter_id = 1 exists in the table of compounds named 'emolecules_sample'. The matched molecules will be written to a table named 'emolecules_sample_filter_id_1'. As you can see, the table is named after the original table name followed by the filter_id that was applied.
Upong depiction of the resulting table, the following molecules can be seen:
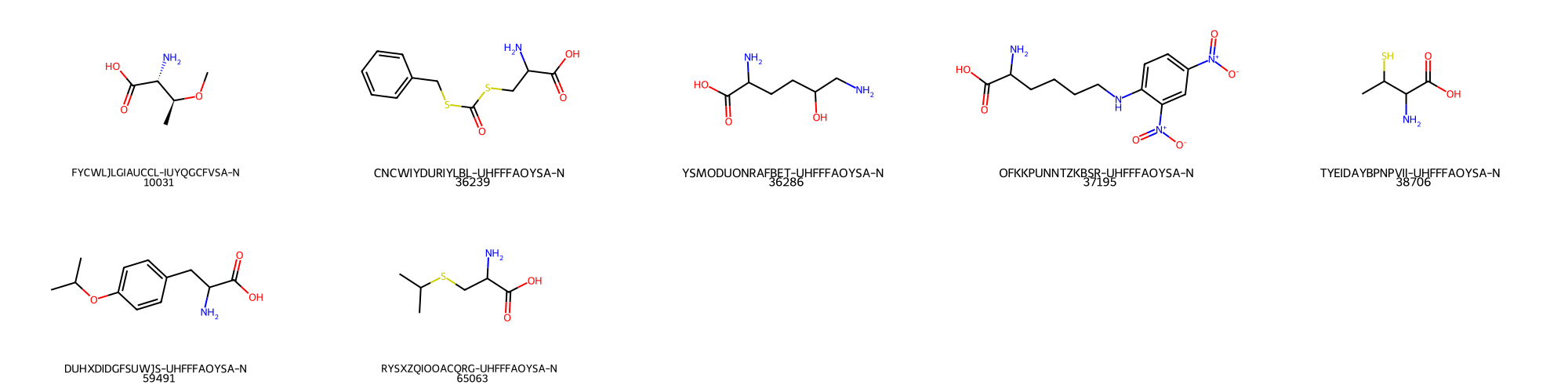
A can be seen, the filtering of molecules containing aminoacids was succesfull.
As expected, custom filters can be added to the default ones provided upon installation of TidyScreen. To perform this, the user should use the add_chem_filters_from_file , which will read a csv file containing the following information:
- Field_1:
filter_name - Field_2:
filter_smarts - Field_3:
user_name
NOTE: continue with example of adding a custom filter for thiols and filtering with 2 ids.
Construction of libraries from databases
Under construction
Exploration of in-house compounds libraries
Under construction
Enumeration of synthetic chemical routes
This approach to the exploration of a chemical is associated to the definition of the type of chemistry that is going to be applied in the research project, both virtually and experimentally. Thus this approach is characterized by the existence of a previous knowledge respect to the synthetic feasibility of the compounds to be explored, including the synthetic versatility of each reaction step that is programmed. Tipically, in this stage there is a close communication between the scientist performing the virtual studies and the organic chemist that is going to synthesize the proposed candidates.
Briefly, the focus of the strategy is centered on one or more synthetic route/s towards the desired candidates, which in turn is related to the type and number of reactants involved in each step of the whole synthetic workflow. Sometimes, upon discussion with organic chemists, even the reactants of a given reaction stage can be indeed obtained through specific synthetic protocols, which further expands the experimental network towards the compounds in study. Following this approach, a massive chemical space with synthetic feasibility can be constructed and afterwards explored.
In short, the wole process is organized as follows:
- Input of reactants into TidyScreen
- Definition of a chemical reaction to transform reactants
- Execution of a chemical reaction pipelline and storage of products
Input of reactants into TidyScreen
Se previous section dealing with the database ingestion and/or .csv file reading.
Definition of a chemical reaction to transform reactants
Chemical reactions can be simulated in silico by using the SMARTS reaction notation, which is implemented in the Python package RDKit, a central component of TidyScreen.
When TidyScreen is first installed, a custom set of reactions are already provided as part of the default reactions_database. It should be noted that this reactions_database belongs to the installed TidyScreen package and is not project specific, granting the possibility of applying common sunthetic procedures (such as chemical protection, deprotection, ionizations, etc) on different projects. Available reactions can be listed by creating a main_database object and afterwards use the list_available_single_reaction method as follows:
...
main_database = ts_cfg.MainDbConfigs()
main_database.list_available_single_reactions()
Upon execution of this script, the following output will be provided showing the built-in chemical reactions:
These are the rections available in the main database
1: Azide_from_alpha_aminoacid - [NX3;H2:1][CX4:2][CX3,H0:3]>>[N-]=[N+]=[NX2;H0:1][CX4:2][CX3,H0:3]
2: Acylation - [CX3:1](=[O:2])[OX2H,OX1-:3].[C:4][O]>>[C:1](=[O:2])[O:3][C:4]
3: CuAAC - [NX1-:1]=[NX2+:2]=[NX2:3].[CX2H1:4]#[CX2H0:5]>>[NX2+0:1]1=[NX2+0:2][N:3]-[C:4]=[C:5]1
4: A3Coupling - [N:1].[CX3H1:2](=[O:3])>>[N:1][C:2][C:4]#[C:5]
5: DIBAL - [CX3:1](=[O:2])[OX2H,OX1-:3]>>[CX3H1:1](=[O:2])
6: Thiolation - [CX3:1](=[O:2])[OX2H,OX1-:3]>>[CX4:1][SX2H]
...
As can be seen , each reaction contains a reaction_id associated to a reaction_name and reaction_smarts.
New reaction types can be added to the default ones included in the default database by using the add_single_reactions_from_file method, which will read a .csv file containing the following information:
- Field_1:
reaction_name - Field_2:
reaction_smarts - Field_3:
user_name
This is the default .csv file installed upon the initial installation.
It should be noted that sometimes the creation of a reaction SMARTS can be tricky in terms of unambiguosly matching the correct chemical groups leading to the desired reaction product. The user should check the correctness of the SMARTS notation to be included in the database, otherwise synthetic artifacts will be included in the working chemical space under construction. To aid in this process, the SMARTS PLUS is a very usefull web application to create, view and check SMARTS pattern and matching.
Execution of a chemical reaction pipeline and storage of products
Under construction